BULLOUS PEMPHIGOID (BP)
BULLOUS PEMPHIGOID (BP) ICD-10: L12.0
• A bullous autoimmune disease usually in elderly patients.
• Pruritic papular and/or urticarial lesions with large tense bullae.
• Subepidermal blisters with eosinophils.
• C3 and IgG at epidermal basement membrane, antibasement membrane IgG autoantibodies in serum.
• Autoantigens are keratinocyte hemidesmosome proteins.
• Therapy includes topical and systemic glucocorticoids and other immunosuppressants.
EPIDEMIOLOGY
AGE OF ONSET Sixty to 80 years. SEX Equal incidence in males and in females. No known racial predilection. INCIDENCE The most common bullous subepidermal autoimmune disease in Europe.
ETIOLOGY AND PATHOGENESIS
Interaction of autoantibody with bullous pemphigoid (BP) antigen (BPAG1 [BP230] and BPAG2 [type XVII collagen]) in hemidesmosomes of basal keratinocytes (Fig. 6-1) is followed by complement and mast cell activation, attraction of neutrophils and eosinophils, and release of multiple bioactive molecules from inflammatory cells.
CLINICAL MANIFESTATION
Often starts with a prodromal eruption (urticarial, papular lesions) and evolves in weeks to months to bullae that may appear suddenly as a generalized eruption. Initially moderate or severe pruritus; later, tenderness of eroded
lesions. No constitutional symptoms, except in widespread, severe disease. SKIN LESIONS Erythematous, papular, or urticarial-type lesions (Fig. 6-13) may precede bullae formation by months. Bullae: small (Fig. 6-13) or large (Fig. 6-14), tense, firm-topped, oval, or round; arise in normal, erythematous, or urticarial skin and contain serous (Fig. 6-14) or hemorrhagic fluid. Localized or generalized, usually scattered but also grouped in arciform and serpiginous patterns. Bullae rupture less easily than in pemphigus, but sometimes large, bright red, oozing, and bleeding erosions occur. Usually bullae collapse and transform into crusts.
Sites of Predilection Axillae; medial aspects of thighs, groins, and abdomen; flexor aspects of forearms; lower legs (often first manifestation); generalized. MUCOUS MEMBRANES Practically only in the mouth (10% to 30%); less severe and painful, and less easily ruptured than in pemphigus (see Section 33).
LABORATORY EXAMINATIONS
DERMATOPATHOLOGY Light Microscopy. Eosinophils aligned at dermal–epidermal junction; neutrophils, eosinophils, and lymphocytes in papillary dermis; subepidermal bulla.
Electron Microscopy Junctional cleavage, that is, split occurs in lamina lucida of basement membrane (see Fig. 6-1). IMMUNOPATHOLOGY Linear IgG deposits along the basement membrane zone. Also C3, which may occur in the absence of IgG. SERUM Circulating antibasement membrane IgG antibodies detected by IIF in 70% of patients. Titers may correlate with the course of disease. Autoantibodies recognize two types of antigens. BPAG1 is a 230-kDa glycoprotein that has high homology with desmoplakin I and is part of hemidesmosomes. BPAG2 is a transmembranous 180-kDa polypeptide (type XVII collagen) (see Fig. 6-1). HEMATOLOGY Eosinophilia (not always).
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
Clinical appearance, histopathology, and immunology permit a differentiation from other bullous diseases (see Table 6-3).
MANAGEMENT
Systemic prednisone with starting doses of 1 to 2 mg/kg continued until no new lesions or itch for 2 weeks. Doxycycline (100 mg BID, +/- nicotinamide), dapsone (50 to 200 mg daily), methotrexate (5 to 10 mg weekly), azathioprine (2.5 mg/kg), and mycophenolate mofetil (2 to 3 mg daily) can be added for corticosteroid-sparing therapy. Topical corticosteroids can also be used, especially in milder cases.
COURSE AND PROGNOSIS
Prognosis is variable, and generally follows a chronic, relapsing course.
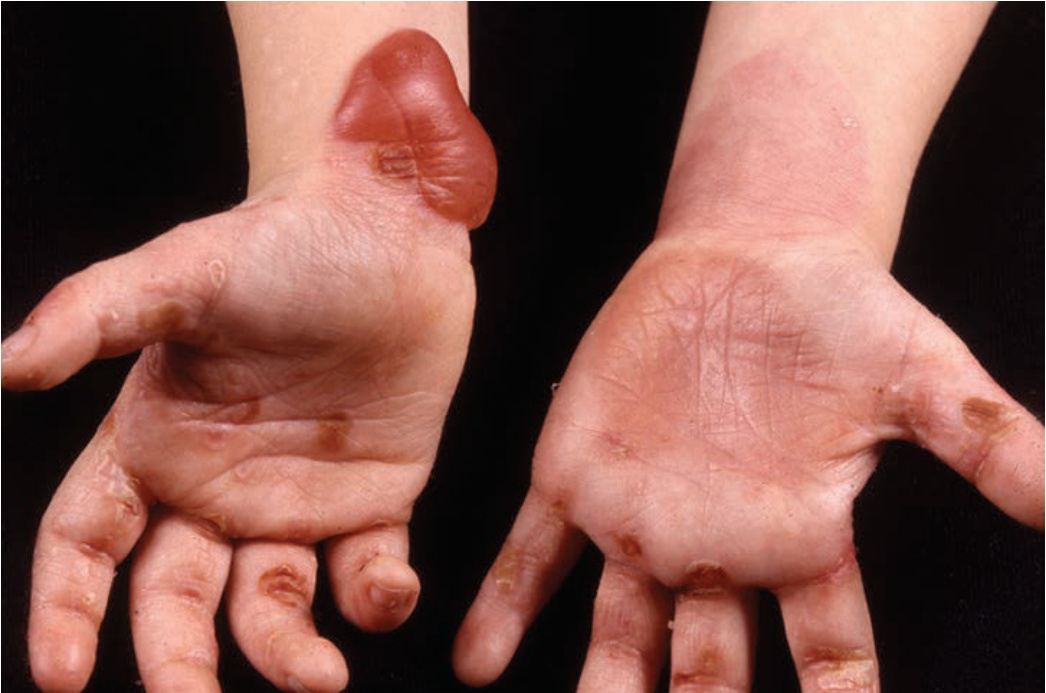
FIGURE 6-1 • Schematic of the components of dermal–epidermal basement membrane and levels of dermal–epidermal separation in hereditary and autoimmune bullous diseases with dermal–epidermal cleavage discussed in this Atlas EBS, epidermolysis bullosa simplex; BP, bullous pemphigoid; PG, pemphigoid gestationis; LAD, linear IgA disease; CP, cicatricial pemphigoid; EBA, epidermolysis bullosa acquisita; DEB, dermolytic epidermolysis bullosa. (Modified with permission from Marinkovich MP. Inherited epidermolysis bullosa. In: Goldsmith LA, Katz SI, Gilchrest BA, et al., eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012, pp. 649–665.)
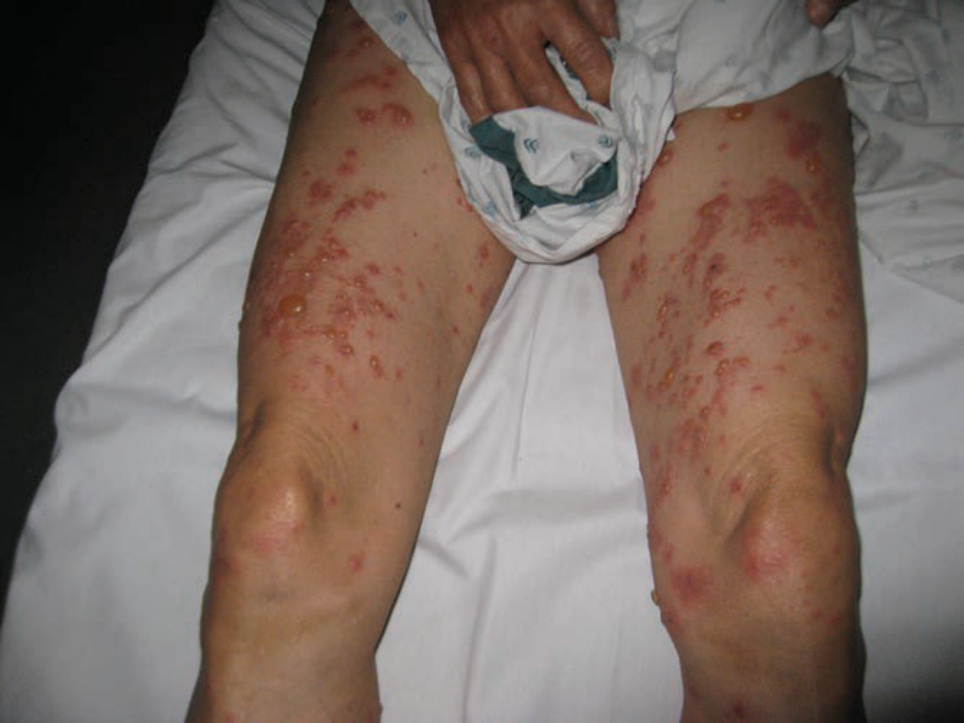
FIGURE 6-13 • Bullous pemphigoid Tense bullae and urticarial plaques on the legs.
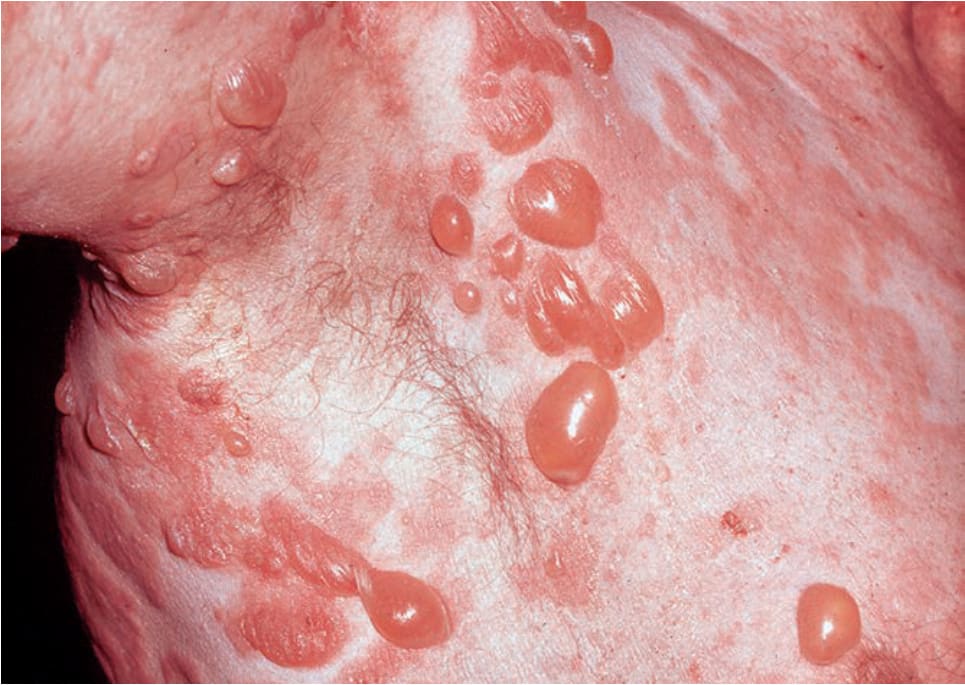
FIGURE 6-14 • Bullous pemphigoid This 77-year-old male has a generalized eruption with confluent urticarial plaques and multiple tense blisters. The condition is severely pruritic.
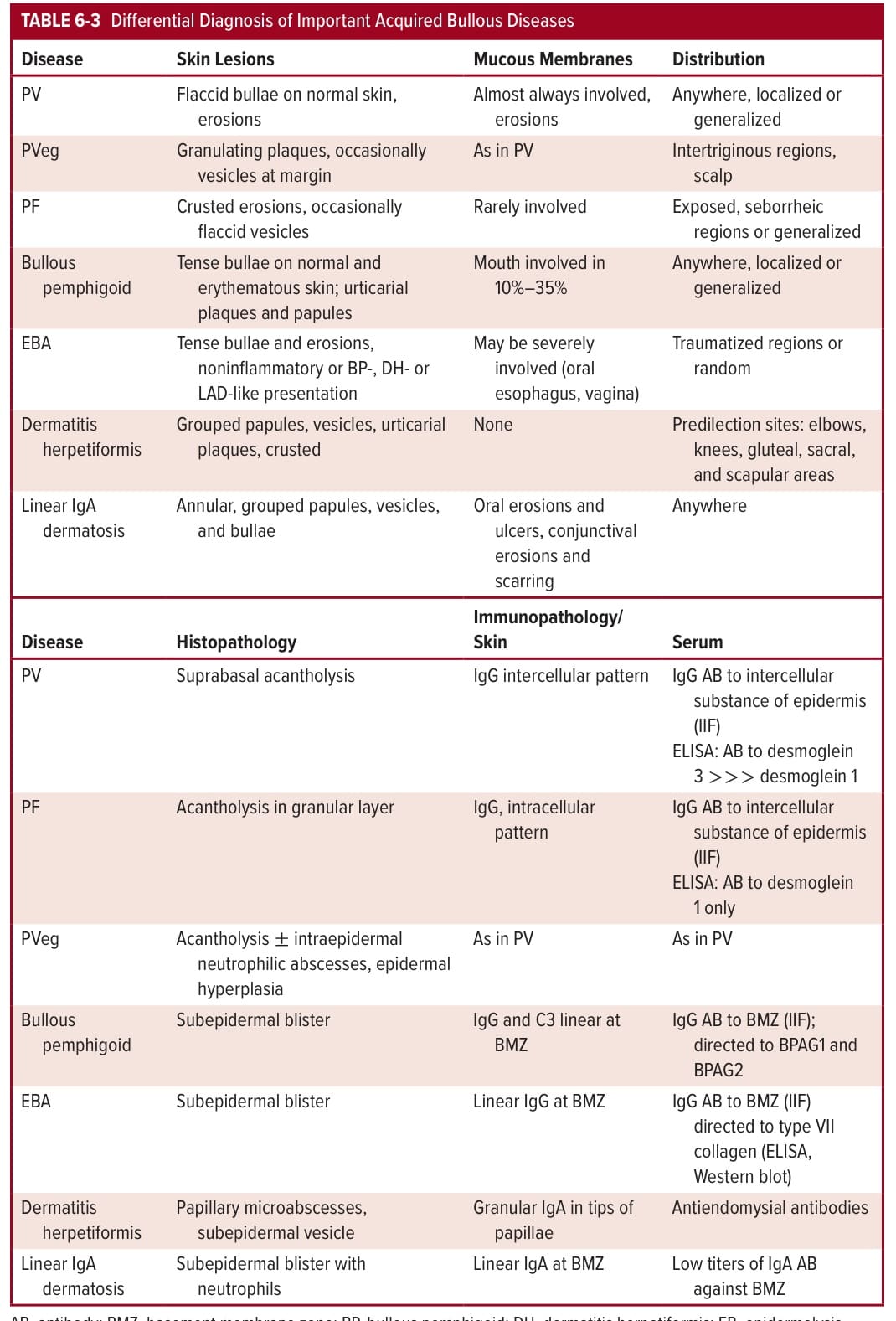
TABLE 6-3 Differential Diagnosis of Important Acquired Bullous Diseases