DERMATOMYOSITIS
DERMATOMYOSITIS ICD-10: M33.0
• DM is a systemic disease belonging to the idiopathic inflammatory myopathies, a heterogeneous group of genetically determined autoimmune diseases targeting the skin and/or skeletal muscles.
• DM is characterized by violaceous (heliotrope) inflammatory changes +/- edema of the eyelids and periorbital area; erythema of the face, neck, and upper trunk; and flat-topped violaceous papules over the knuckles.
• It is associated with polymyositis, interstitial pneumonitis, and myocardial involvement.
• There is also a DM without myopathy (amyopathic DM) and polymyositis without skin involvement.
• Juvenile DM runs a different course and is associated with vasculitis and calcinosis.
• Adult-onset DM may be associated with internal malignancy.
• Prognosis is guarded.
EPIDEMIOLOGY AND ETIOLOGY
RARE Incidence >6 cases per million, but this is based on hospitalized patients and does not include individuals without muscle involvement. Juvenile and adult (>40 years) onset. ETIOLOGY Unknown. In persons >55 years of age, may be associated with malignancy. CLINICAL SPECTRUM Ranges from DM with only cutaneous inflammation (amyopathic DM) to polymyositis with only muscle inflammation. Cutaneous involvement occurs in 30% to 40% of adults and 95% of children with DM/polymyositis. For classification, see Table 14-2.
CLINICAL MANIFESTATION
SYMPTOMS + PHOTOSENSITIVITY Manifestations of skin disease may precede myositis or vice versa; often, both are detected at the same time. Muscle weakness, difficulty in rising from supine position, climbing stairs, raising arms over head, or turning in bed. Dysphagia; burning and pruritus of the scalp. SKIN LESIONS Periorbital heliotrope (reddish purple) flush, usually associated with some degree of edema (Fig. 14-22). May extend to involve scalp (+ nonscarring alopecia), entire face, upper chest, and arms (Fig. 14-23A).
Dermatomyositis (DM)
● Adult onset
● Classic DM: alone; with malignancy; as part of an overlap connective tissue disorder
● Clinically amyopathic DM: amyopathic DM: hypomyopathic DM
● Juvenile onset
● Classic DM
● Clinically amyopathic DM: amyopathic DM; hypomyopathic DM
Polymyositis (PM)
● PM alone
● PM as part of an overlap connective tissue disorder
● PM associated with internal malignancya
Inclusion body myositis
Other clinical–pathologic subgroups of myositis
● Focal myositis
● Proliferative myositis
● Orbital myositis
● Eosinophilic myositis
● Granulomatous myositis
aAlthough population-based European studies have now clearly confirmed that adult-onset classic DM is associated with a significant risk for internal malignancy, the evidence that such a relationship exists for PM, is much weaker.
In addition, papular dermatitis with varying degrees of violaceous erythema in the same sites. Flat-topped, violaceous papules (Gottron papules) with various degrees of atrophy on the nape of the neck and shoulders and over the knuckles and interphalangeal joints (Fig. 14-23B). Note: In lupus, lesions usually occur in the interarticular region of the fingers (see p. 335). Periungual erythema with telangiectasia, thrombosis of capillary loops, and infarctions. Lesions over elbows and knuckles may evolve into erosions and ulcers that heal with stellate scarring (particularly in juvenile DM with vasculitis). Long-lasting lesions may evolve into poikiloderma (mottled discoloration with red, white, and brown) (Fig. 14-24). Calcification in subcutaneous/fascial tissues common later in the course of juvenile DM (Fig. 14-25), particularly around the elbows, trochanteric, and iliac region (calcinosis cutis); may evolve into calcinosis universalis. MUSCLE ± Muscle tenderness, ± muscle atrophy. Progressive muscle weakness affecting proximal/limb girdle muscles.
A
B
Occasional involvement of facial/bulbar, pharyngeal, and esophageal muscles. Deep tendon reflexes within normal limits. OTHER ORGANS Interstitial pneumonitis, cardiomyopathy, arthritis, particularly in juvenile DM (20% to 65%). DISEASE ASSOCIATION Patients >50 years of age with DM have a higher than expected risk for malignancy, particularly ovarian cancer in females. Also carcinoma of the breast, bronchopulmonary, and GI tract.
LABORATORY EXAMINATIONS
CHEMISTRY Elevation of creatine phosphokinase (65%), aldolase (40%), lactate dehydrogenase, and glutamic oxaloacetic transaminase. AUTOANTIBODIES Autoantibodies to 155 kDa and/or Se in 80% to 140%, kDa in 58%, and to Jo-1 in 20% and to (low specificity) antinuclear antibodies (ANA) in 40%. Anti-Mi-2 (classic DM); Anti-TIF-1 (related to cancer risk;
juvenile disease), Anti-NXP2 (severe muscle disease, calcinosis), Anti-MDA5 (progressive interstitial lung disease; amyopathic). URINE Elevated 24-hour creatine excretion (>200 mg/24 hours). ELECTROMYOGRAPHY Increased irritability on insertion of electrodes, spontaneous fibrillations, pseudomyotonic discharges, and positive sharp waves. MRI MRI of muscles reveals focal lesions. ECG Evidence of myocarditis; atrial and ventricular irritability; atrioventricular block. X-RAY Chest ± interstitial fibrosis. Esophagus: reduced peristalsis. PATHOLOGY Skin Flattening of epidermis, hydropic degeneration of basal cell layer, edema of upper dermis, scattered inflammatory infiltrate, PAS-positive fibrinoid deposits at dermal–epidermal junction, accumulation of acid mucopolysaccharides in dermis (all these are compatible with DM but are not diagnostic).
Muscle Biopsy shoulder/pelvic girdle; one that is weak or tender. Histology—segmental necrosis within muscle fibers with loss of cross striations; myositis. Vasculitis is seen in juvenile DM. MALIGNANCY SCREENING Consider stool occult blood testing, mammography and pelvic ultrasound, CT of chest abdomen and pelvis; age appropriate screening.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
Skin signs plus proximal muscle weakness with two of three laboratory criteria, that is, elevated serum “muscle enzyme” levels, characteristic electromyographic changes, and diagnostic muscle biopsy. Differential diagnosis is to lupus erythematosus, mixed connective tissue disease, steroid myopathy, trichinosis, and toxoplasmosis.
COURSE AND PROGNOSIS
Prognosis guarded but with treatment, it is relatively good except in patients with malignancy and those with pulmonary
involvement. With aggressive immunosuppressive treatment, the 8-year survival rate is 70% to 80%. A better prognosis is seen in individuals who receive early systemic treatment. The most common causes of death are malignancy, infection, cardiac, and pulmonary disease. Successful treatment of an associated neoplasm is often followed by improvement/ resolution of DM.
MANAGEMENT
PREDNISONE 0.5 to 1 mg/kg body weight per day. Taper when “muscle enzyme” levels approach normal. Best if combined with azathioprine, 2 to 3 mg/kg per day. Note: Steroid myopathy may occur after 4 to 6 weeks of therapy. ALTERNATIVES Methotrexate, hydroxychloroquine, azathioprine, cyclophosphamide, cyclosporine, antitumor necrosis factor (TNF) α agents. High-dose IV immunoglobulin bolus therapy (2 g/kg body weight given over 2 days) at monthly intervals spares glucocorticoid doses to achieve or maintain remissions. Janus kinase inhibitors (JAK) can be helpful in recalcitrant disease. Rituximab has been studied in myositis.
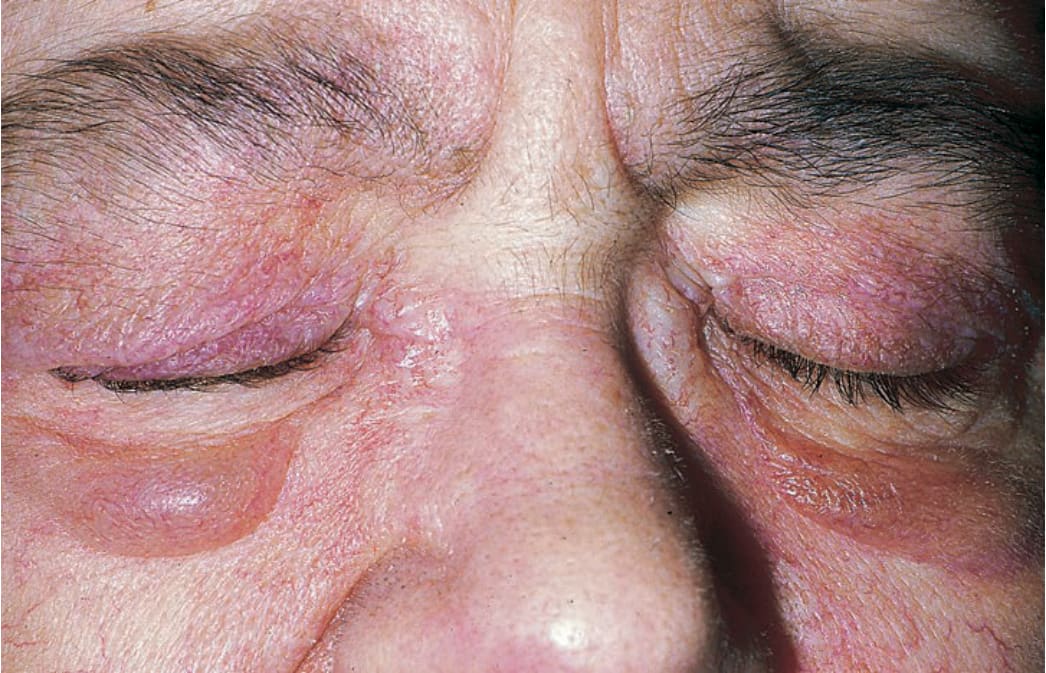
FIGURE 14-22 • Dermatomyositis Heliotrope (reddish purple) erythema of upper eyelids and edema of the lower lids. This 55-year-old female had experienced severe muscle weakness of the shoulder girdle and presented with a lump in the breast that proved to be carcinoma.
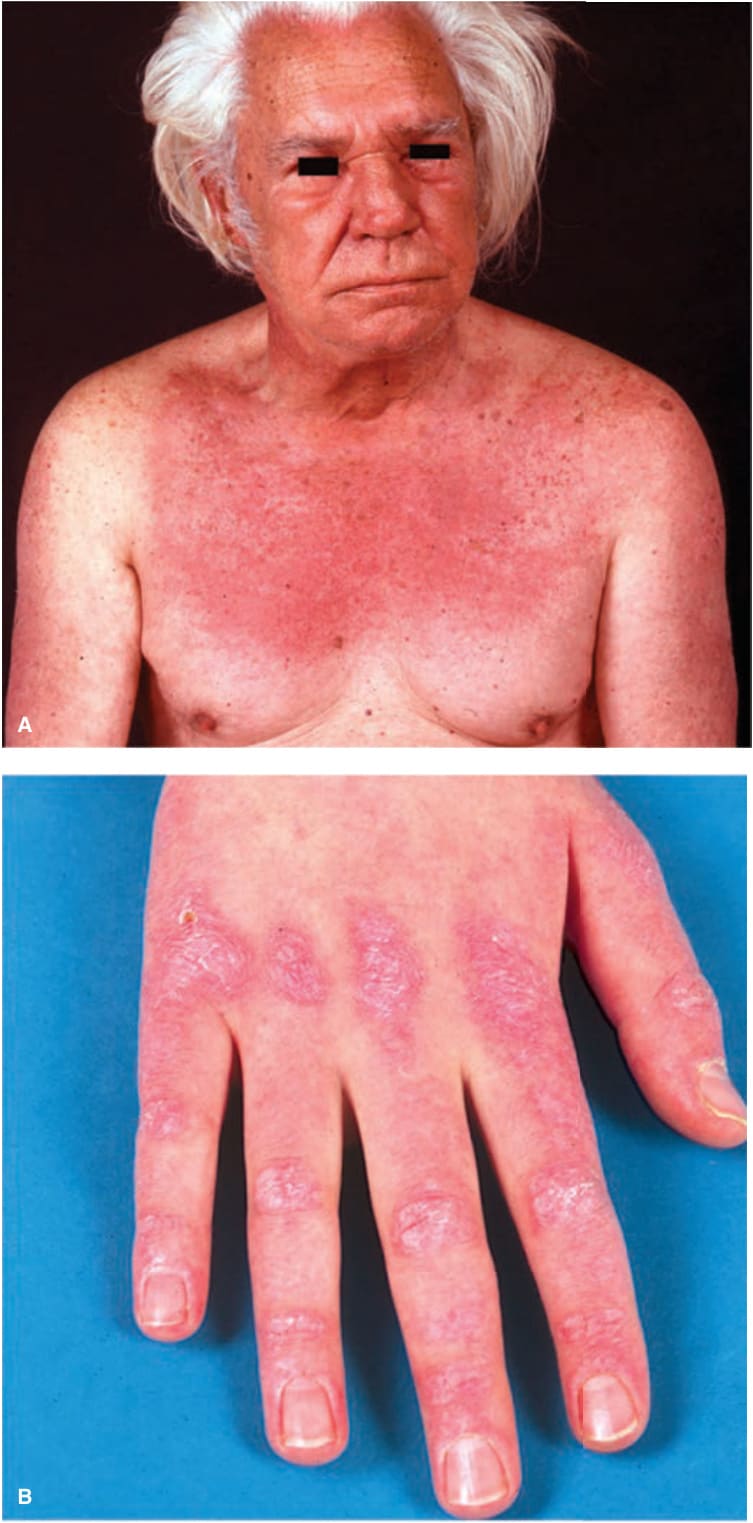
FIGURE 14-23 • Dermatomyositis (A) Violaceous erythema and edema on the face, particularly in the periorbital and malar regions and on the chest. The patient could barely lift his arms and could not climb stairs. (B) Violaceous erythema and Gottron papules on the dorsa of the hands and fingers, especially over the interphalangeal joints. Periungual erythema and telangiectasias.
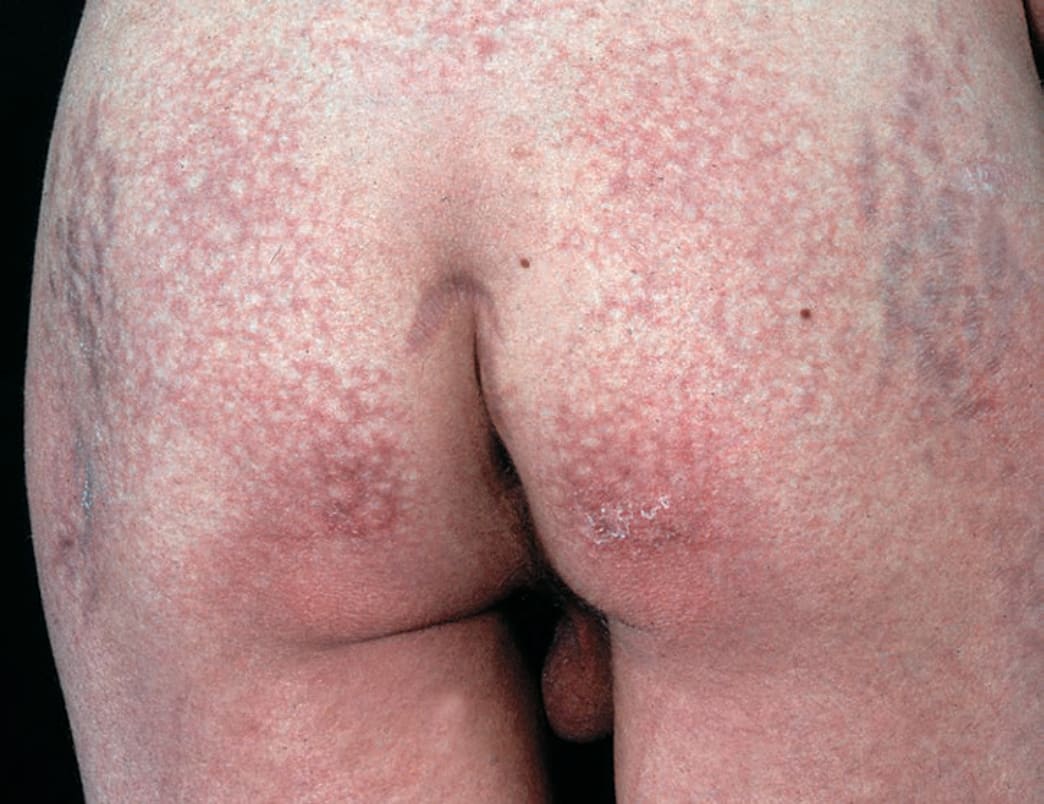
FIGURE 14-24 • Dermatomyositis, juvenile onset, poikiloderma There is mottled, reticular brownish pigmentation and telangiectasia plus small white scars. Note striae on trochanteric areas caused by systemic glucocorticoid therapy.
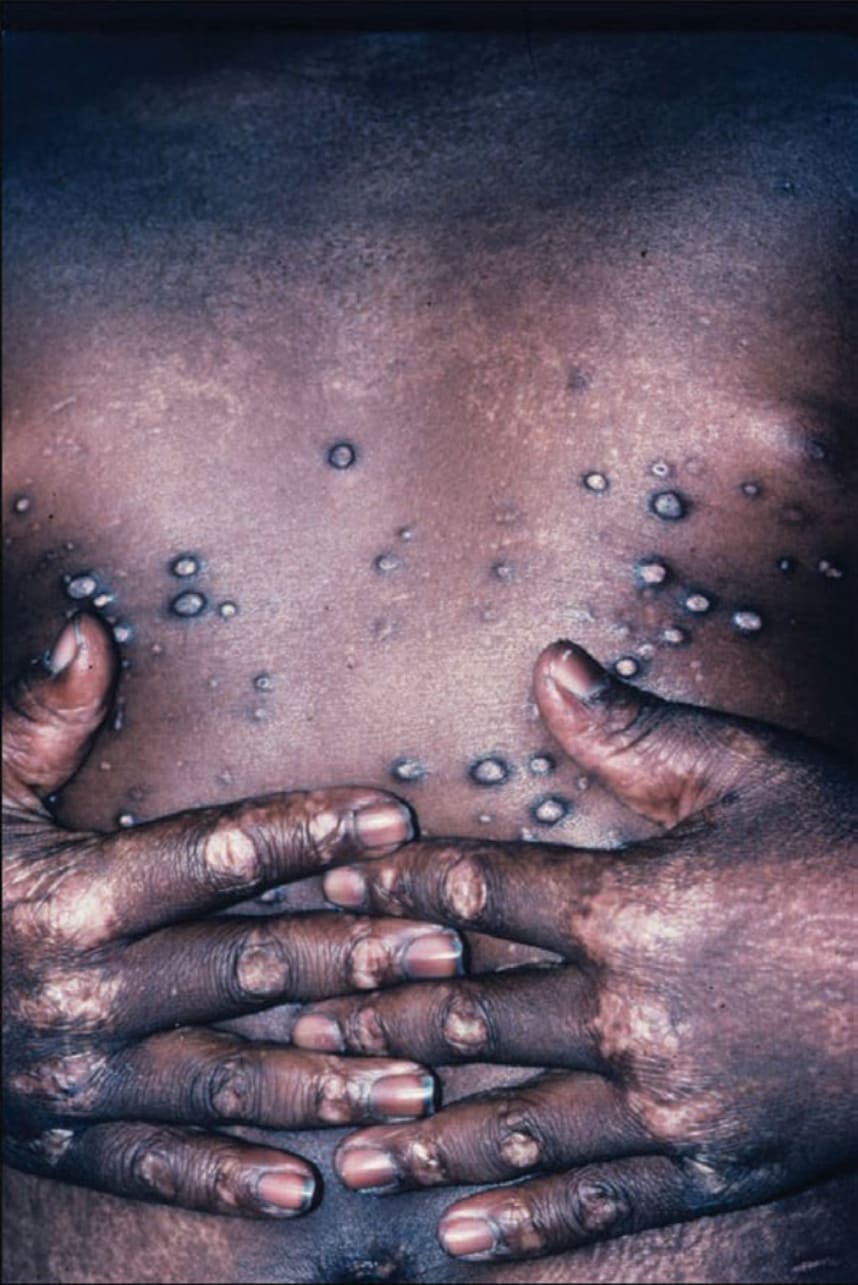
FIGURE 14-25 • Dermatomyositis Calcinosis. There are several hard nodules, which may later ulcerate and reveal a chalk white mass at the base. Upon squeezing, they will exude a white paste. (Used with permission of Dr. Kenneth Greer.)
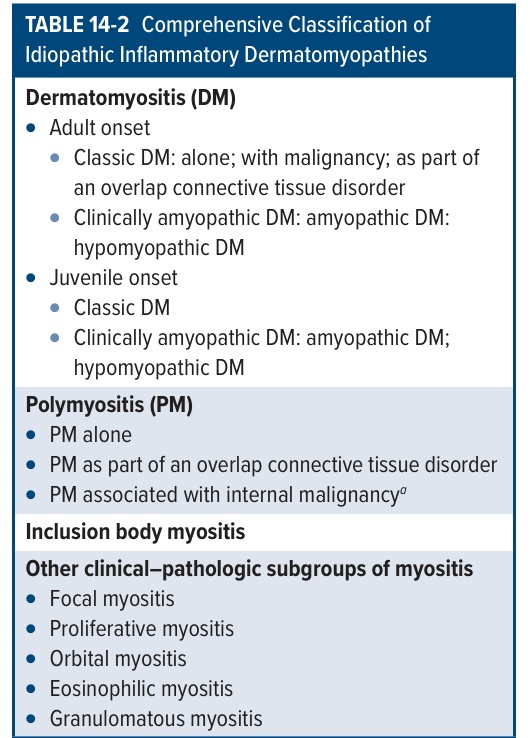
TABLE 14-2 Comprehensive Classification of Idiopathic Inflammatory Dermatomyopathies